|
|
|
|
|
|
|
|
Stichwortverzeichnis: |
|
|
|
|
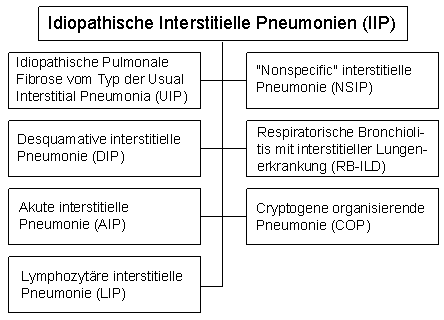
1. Allgemeine CharakterisierungEine große Zahl verschiedenartiger chronischer Krankheiten des Lungenparenchyms, die in einer Fibrose enden, werden auch unter dem Begriff "chronisch interstitielle Pneumonien" oder "interstitielle Lungenkrankheiten" oder "Lungengerüstkrankheiten" zusammengefaßt. Im Englischen heißen sie "Diffuse Parenchymal Lung Diseases" (DPLD) oder "Interstitial Lung Diseases" (ILD). Ursachen der Fibrose sind: - überwiegend unbekannte Äthiologie ("idiopathisch", "cryptogen"),
- idiopathische pulmonale Fibrose (IPF) mit dem Bild einer UIP (Usual Interstitial Pneumonia) - darin finden sich die meisten idiopathischen Fibrosen. Seltener sind die Folgenden:
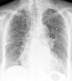
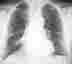
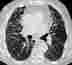
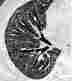
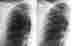
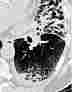
Im Röntgenübersichtsbild kann ein feinstreifig-retikuläres Muster, daß von einer diffusen Transparenzminderung überlagert wird, erscheinen:
Das Thoraxübersichtsbild ist jedoch häufig unauffällig. Deshalb ist bei interstitiellen Erkrankungen die Computertomographie (HRCT) die Methode der Wahl:
|
|
|
|
2. Allergische
Alveolitis
(
Hypersensitivitätspneumonie) - Amiodaron (Cordarex®), oder durch organisches Material. Darunter finden sich zahlreiche Allergene, die oft auch berufstypische Hypersensitivitätspneumonien verursachen: - schimmeliges Heu (Farmerlunge) Einige der als Berufskrankheiten anerkannten Lungenkrankheiten sind allergischer Genese. Nach dem auslösendes Ereignis (Trauma, Schock, immunologische Schädigung) treten Veränderungen auf, die pathophysiologisch in 3 Stadien unterteilt werden:
Klinisch treten 6-8 Stunden nach massiver Allergeneinwirkung (Stall ausmisten, Heu verladen) rezidivierendes Fieber, Dyspnoe und Husten auf. Bei chronischer Einwirkung (Klimaanlagen, Wellensittighaltung) bestehen Mattigkeit und Gewichtsverlust wie bei einem okkulten Tumor. Die Lungenfunktion zeigt eine restriktive Ventilationsstörung. Medikamentös, durch Pilze oder Parasiten induzierte Infiltrate, oder Infiltrate bei rheumatoiden Erkrankungen sind oft von einer Eosinophilie begleitet (siehe unten). Bei der Farmerlunge handelt es sich um eine allergische Reaktion auf inhalierte Microsporen aus feuchtem Heu oder Luftbefeuchtern. Bei schlecht gewarteten Klimaanlagen kann die "Farmerlunge" also auch in der Stadt vorkommen. Die akute Entzündung klingt ab, wenn die Exposition beendet wird. Nach ca. 5 wiederholten Erkrankungen nimmt die Wahrscheinlichkeit einer Lungenfibrose stark zu. Neben der allergischen liegt auch eine toxische Wirkung durch bakterielle Endotoxine vor. Computertomographische Zeichen:
Differentialdiagnostisch abzugrenzen ist die UIP (Usual Interstitial Pneumonia) mit Milchglastrübung, Linienzeichnung, Wabenlunge, d.h. alle Stadien einer entzündlichen interstitiellen Reaktion nebeneinander, aber keine unscharfen Knötchen.
|
|
|
|
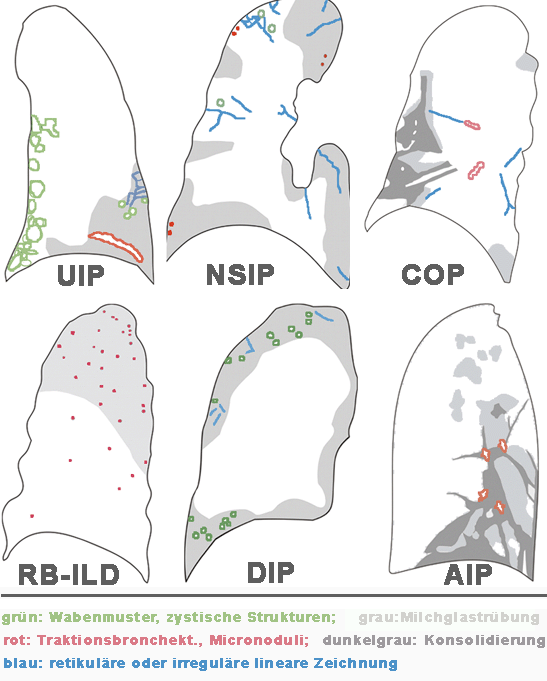
Klassifikation idiopathischer interstitiellerPneumonien
Klinik radiologi-sche Zeichen Vertei-lungs-muster im CT Zeichen im CT Differential-diagnose IPF/UIP basale und periphere Netzzeich-nung;
Schrumpfung basal und peripher sub-pleural Wabenmuster, Bronch-ektasen, strukturelle
Deformität, fokale Milch-glastrübung Asbestose, Kollagenose,
Hypersensitivi-tätspneumonie, Sarkoidose DIP Milchglas-trübung basal und peripher Milchglastrübung, retikuläre
Zeichnung RB-ILD, Hypersensitivi-tätspneumonie, PcP,
Sarkoidose RB-ILD Bronchus-wandver-dickung,
Milchglas-trübung diffus Bronchuswandver-dickung, fleckige
Milchglastrübung, zentrilobuläre
Knötchen DIP, NSIP, Hypersensitivi-tätspneumonie AIP progressive diffuse Milch-glastrübung,
Konsoli-dierung diffus Milchglastrübung, Konsolidierung, oft mit
lobulären Aussparungen,
Traktionsbronch-ektasen orthostatisches Ödem, Pneumonie, akute
eosinophiele Pneumonie COP fleckige beidseitige Konsolidierungen peribron-chial, sub-pleural fleckige Konsolidie-rungen, und/oder
Knötchen Infektion, NSIP, Vaskulitis, Sarkoidose,
Alveolarzell-Ca, Lymphom, eosinophiele
Pneumonie NSIP Milchglas-trübung, retikuläre
Ver-dichtungen basal und peripher sub-pleural, symme-trisch Milchglastrübung, Konsolidierung,
irreguläre Bänder IPF/UIP, DIP, COP,
Hypersensitivi-tätspneumonie LIP retikuläre Verdichtungen, Knötchen diffus Milchglastrübung zentrilobuläre
Knötchen, broncho-vaskuläre- und
Septenverdickung Sarkoidose, Lymphangiosis carcinomatose,
Langerhanszell-Histiozytose Abkürzungen:
IPF/UIP idiopathische pulmonale Fibrose mit dem Bild
einer UIP (gewöhnliche interstitielle
Pneumonie) COP kryptogene organisierende Pneumonie (früher
BOOP) DIP desquamative interstitielle Pneumonie NSIP unspezifische interstitielle Pneumonie R-BILD bronchiolitis respiratorii interstitielle
Pneumonie LIP lymphozytäre interstitielle Pneumonie AIP akute interstitielle Pneumonie . . |
|
|
|
AIP: Im Gegensatz zur UIP ist das computertomographische Bild der AIP homogen:
UIP: Die CT ist sehr spezifisch. Nachgewiesen wird die "temporäre Heterogenität" (verschiedene Erscheinungsformen nebeneinander, bevor das homogene Bild einer Fibrose entsteht), das sind:
NSIP: HRCT-morphologisch steht die NSIP zwischen der UIP (periphere basale Fibrose) und DIP (basale periphere Milchglastrübung).
DIP :
RB-ILD (Respiratory
Bronchiolitis-associated Interstitial Lung Disease) ist eine
durch Rauchen hervorgerufenesehr seltene interstitielle
Lungenkrankheit. Andere, durch Rauchen verursachte
interstitielle Lungenkrankheiten sind DIP, Langerhanszell
Histiozytose und IPF. Das histologische und
computertomographische Bild der RB-ILD ähnelt oft der
DIP. Meist besteht ein zentriazinäres Emphysem.
|
|
|
|
Die anderen 50 % sind idiopathisch oder cryptogen (dann COP). Die COP ist eine Ausschlußdiagnose. Vor Cortisontherapie müssen evtl. bekannte Ursachen ausgeschlossen werden, wie Infektionskrankheiten. Meist sind Frauen im mittleren Alter betroffen. Klinisch bestehen Dyspnoe und trockener Husten sowie geringes Fieber über 4 bis 10 Wochen. Die Lungenfunktionswerte zeigen eine geringe Restriktion. Die Erkrankung spricht auf Cortison gut an. Pathohistologisch finden sich in den Bronchiolen Pfropfen von proliferierenden Fibroblasten. In der umgebenden Lunge bestehen Pneumonieherde, die in Organisation übergehen. Computertomographische Befunde: Typisch für die COP sind die Konsolidierungen und Milchglastrübung bds.. basal ohne Fibrose. Diese Konfiguration spricht gegen eine UIP oder DIP. Die Infiltrate treten basal auf. Dies unterscheidet sie von den mehr kranial lokalisierten Infiltraten des eosinophilen Granuloms.
Therapie der Idiopathischen Interstitiellen Pneumonien:
|
|
|
|
4. SarkoidoseSynonym: Morbus Besnier-Boeck-Schaumann. Es handelt sich um eine Multisystemerkrankung mit nichtverkäsenden epitheloidzelligen Granulomen. Die Genese ist unbekannt. Man vermutet eine genetische, bedingt erworbene Immunopathie. Neben Lunge und hilären Lymphknoten können auch alle andere Organe befallen werden. Klinisch manifestiert sich die Krankheit
in frühen und mittleren Lebensabschnitten, Frauen sind
häufiger betroffen als Männer, Schwarze mehr als
andere Rassen. Hohe Krankheitszahlen bestehen in den USA
(schwarze Bevölkerung), in Schweden und Island. Die
Prävalenz in der BRD beträgt 16 auf 100.000
Einwohner, die Mortalität ca. 1:1.000.000
Einwohner.
In der Lungenfunktion besteht eine Restriktion, die bei
Fibrose in eine Obstruktion und terminal in eine
respiratorische Insuffizienz bei Cor pulmonale
übergeht. Die Diagnosesicherung erfolgt histologisch
durch Bronchoskopie, da differentialdiagnostisch bei
bihilären Lymphomen auch an ein Hodgkin-Lymphom gedacht
werden muß. Röntgentyp I hilärer oder medistinaler Befall Bewußt wird von Röntgentypen und nicht von Stadien, die von 1-3 fortschreiten könnten, gesprochen. Lediglich der Typ IV ist ein irreversibles Endstadium. Röntgentyp I kommt am häufigsten vor, Röntgentyp I und II haben die höchste Spontanremissionsrate. Die Klassifikation stützt sich auf das Thoraxübersichtsbild. Trotzdem wird die Einteilung zunehmend auch für CT verwendet - obwohl die Ergebnisse nicht vergleichbar sind. Die computertomographisch dargestellte
Milchglastrübung als Zeichen der akuten Erkrankung und
die Knoten können sich zurückbilden, nicht aber
die Fibrose. Die kleinen Granulome sind zunächst als
einzelne Knötchen sichtbar. Wenn sie sich zahlreicher
entlang den bronchovaskulären Strukturen aufreihen,
bilden sie im Röntgenbild eine Netzzeichnung. Zur Typisierung wird die Computertomographie herangezogen, da sowohl hiläre wie auch intrapulmonale Prozesse genauer bestimmt werden können als mit der konventionellen Radiologie - sie ist aber nicht mit den Ergebnissen von Thoraxübersichtsbildern vergleichbar. Die Krankheitsaktivität wird anhand der Klinik, des Röntgens und der Lungenfunktion bestimmt, seltener durch BAL, CD4/CD8-Zellquotienten oder Serum-ACE (Angiotensin I converting enzyme). Die Aktivitätsbestimmung durch Szintigraphie wird nicht mehr durchgeführt. Neuerdings wird versucht, die Aktivität interstitieller Erkrankungen mittels MR (T1-Bild) zu bestimmen. Röntgenzeichen im Thoraxübersichtsbild:
Röntgenzeichen im Computertomogramm:
- Lymphangiosis
carcinomatosa |
|
|
|
5. Langerhans-Zell-HistiozytoseDie Langerhans-Zell-Histiozytose, früher auch Eosinophiles Granulom oder nur Histiozytosis X genannt, tritt zu 95% bei schweren Rauchern auf. Es ist eine Granulomatose aufgrund einer erhöhten Aktivierung lokaler epithelständiger Langerhans-Zellen. Dies kann in allen Organen auftreten, zu ca. 70% aber in der Lunge. Klinisch treten Husten, Belastungsdyspnoe, Thoraxschmerzen, Fieber und Gewichtsverlust auf. Röntgenzeichen im HRCT:
Differentialdiagnostisch werden im frühen Stadium meist Metastasen vermutet. Strenge Nikotinabstinenz kann zur vollständigen Regression der noch nicht narbigen Veränderungen führen. |
|
|
|
6. Differentialdiagnose diffuser infiltrativer Lungenkrankheiten in der hochauflösenden Computertomographie (HRCT)Siehe auch die Kurzfassung "Knötchen im HRCT"
für "Handhelds" |
|
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|



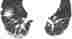














{kind=link}